Welcome to CYANUS: CYtometry ANalysis Using Shiny
Recently, high-dimensional time-of-flight mass cytometry (CyTOF)
has emerged with the ability to identify more than 40 parameters simultaneously.
Traditional flow cytometry would require multiple tubes with different
antibody panels to cover the same number of markers. Consequently,
CyTOF experiments are becoming a powerful tool to unveil new cell subtypes,
functions, and biomarkers in many fields, e.g. the discovery of disease-associated
immunologic changes in cancer.
In order to facilitate the analysis of CyTOF data for biologists and physicians, a clear, understandable and user-friendly pipeline is needed.
Here, we integrated the methods from the CATALYST package for preprocessing, visualization and clustering. For differential abundance detection, we included the diffcyt methods diffcyt-DA-edgeR, diffcyt-DA-voom and diffcyt-DA-GLMM.
However, many experiments aim to detect differential states within cell populations between samples in different conditions. For this, we integrated the published methods diffcyt-DS-limma, diffcyt-DS-LMM, CytoGLMM and CytoGLM. Additionally, we performed a comprehensive analysis of these existing methods and novel approaches published in Briefings in Bioinformatics. Since the Wilcoxon rank-sum test and the t-test on sample medians, as well as our novel method CyEMD performed well, we made them available in this interface.
In order to facilitate the analysis of CyTOF data for biologists and physicians, a clear, understandable and user-friendly pipeline is needed.
Here, we integrated the methods from the CATALYST package for preprocessing, visualization and clustering. For differential abundance detection, we included the diffcyt methods diffcyt-DA-edgeR, diffcyt-DA-voom and diffcyt-DA-GLMM.
However, many experiments aim to detect differential states within cell populations between samples in different conditions. For this, we integrated the published methods diffcyt-DS-limma, diffcyt-DS-LMM, CytoGLMM and CytoGLM. Additionally, we performed a comprehensive analysis of these existing methods and novel approaches published in Briefings in Bioinformatics. Since the Wilcoxon rank-sum test and the t-test on sample medians, as well as our novel method CyEMD performed well, we made them available in this interface.
Welcome to CYANUS: CYtometry ANalysis Using Shiny
Recently, high-dimensional time-of-flight mass cytometry (CyTOF)
has emerged with the ability to identify more than 40 parameters simultaneously.
Traditional flow cytometry would require multiple tubes with different
antibody panels to cover the same number of markers. Consequently,
CyTOF experiments are becoming a powerful tool to unveil new cell subtypes,
functions, and biomarkers in many fields, e.g. the discovery of disease-associated
immunologic changes in cancer.
In order to facilitate the analysis of CyTOF data for biologists and physicians, a clear, understandable and user-friendly pipeline is needed.
Here, we integrated the methods from the CATALYST package for preprocessing, visualization and clustering. For differential abundance detection, we included the diffcyt methods diffcyt-DA-edgeR, diffcyt-DA-voom and diffcyt-DA-GLMM.
However, many experiments aim to detect differential states within cell populations between samples in different conditions. For this, we integrated the published methods diffcyt-DS-limma, diffcyt-DS-LMM, CytoGLMM and CytoGLM. Additionally, we performed a comprehensive analysis of these existing methods and novel approaches published in Briefings in Bioinformatics. Since the Wilcoxon rank-sum test and the t-test on sample medians, as well as our novel method CyEMD performed well, we made them available in this interface.
In order to facilitate the analysis of CyTOF data for biologists and physicians, a clear, understandable and user-friendly pipeline is needed.
Here, we integrated the methods from the CATALYST package for preprocessing, visualization and clustering. For differential abundance detection, we included the diffcyt methods diffcyt-DA-edgeR, diffcyt-DA-voom and diffcyt-DA-GLMM.
However, many experiments aim to detect differential states within cell populations between samples in different conditions. For this, we integrated the published methods diffcyt-DS-limma, diffcyt-DS-LMM, CytoGLMM and CytoGLM. Additionally, we performed a comprehensive analysis of these existing methods and novel approaches published in Briefings in Bioinformatics. Since the Wilcoxon rank-sum test and the t-test on sample medians, as well as our novel method CyEMD performed well, we made them available in this interface.
Why is there a new logo?
Our old logo resembled the flower Centaurea cyanus, commonly known as cornflower.
Its strong blue color is utilized as pigment for food and drinks and its leaves can be consumed as tea.
In the past, it has also been used in herbal medicine.
It has recently come to our attention that the flower also has a strong symbolic value. It is, e.g., the national flower of Estonia. In France, it is a symbol for veterans, victims of war, widows, and orphans. In Belgium, Sweden, and Finland, it has been used as symbol for liberal parties. In Germany and Austria, however, it has been used by the Nazis and other nationalist, antisemitic movements.
We explicitly distance ourselves from any connection to this symbolic use and have changed our logo and design.
It has recently come to our attention that the flower also has a strong symbolic value. It is, e.g., the national flower of Estonia. In France, it is a symbol for veterans, victims of war, widows, and orphans. In Belgium, Sweden, and Finland, it has been used as symbol for liberal parties. In Germany and Austria, however, it has been used by the Nazis and other nationalist, antisemitic movements.
We explicitly distance ourselves from any connection to this symbolic use and have changed our logo and design.
Dependencies
Loading...
Get Started
You can upload your own files in the FCS format or use an example dataset that we provide.
Loading...
Data preprocessing
Preprocessing is essential in any mass cytometry analysis process. Usually, the raw marker intensities ready by a cytometer have stronly skewed distributions with varying of expression, thus making it difficult to distinguish between the negative and positive cell populations. The marker intensities are commonly transformed using arcsinh (inverse hyperbolic sine) to make the distributions more symmetric and to map them to a comparable range of expression.
This step also includes some simple visualization plots to verify whether the data represents what we expect, for example, whether samples that are replicates of one condition are more similar and are distinct from samples from another condition. Depending on the situation, one can then consider removing problematic markers or samples from further analysis.
Choose Cofactor for Arcsinh Transformation
No Transformation Applied
You should transform your raw cytometry data.
You can ignore this warning if you supplied already transformed data.
Downsampling
Selecting Samples and Patients
Column Reordering of Metadata
Data Visualization
Here, you can apply a dimensionality reduction method to your data and visualize its results. The following methods are currently supported:
- UMAP : Uniform Manifold Approximation and Projection. Strong mathematical background, results don't vary very much (in comparison to e.g. t-SNE), State-of-the-Art nonlinear visualization method.
- T-SNE: T-distributed stochastic neighbour embedding. Results vary more because of random initialization. Still very popular. Nonlinear method.
- PCA: Principal Component Analysis. The oldest dimensionality reduction technique (1901) but deterministic and very fast. Still a standard method though outperformed by other dimensionality reduction methods. Linear method.
- MDS : Multidimensional scaling. Uses pairwise distances. Linear method.
- Diffusion map: Robust to noise perturbation. Nonlinear method.
- Isomap: Extends MDS by incorporating geodesic distances imposed by a weighted graph. Nonlinear method.
Run the dimensionality reduction with the markers that capture best how dissimilar your cells are. You can color and facet by any marker expression, condition, sample and clustering you wish afterwards.
Clustering
Detect and define cell populations for further downstream analysis via
FlowSOM clustering &
ConsensusClusterPlus meta-clustering:
First, group cells into clusters with FlowSOM using self-organizing map clustering and minimal spanning trees, and subsequently perform meta-clustering with ConsensusClusterPlus which determines cluster count and membership by stability evidence.
A self-organizing map (SOM) is an artificial neural network that reflects the topological information of the input in lower dimensions.
Meta clustering is carried out based on a consensus hierarchical method. By re-sampling from the original data and clustering the samples, an agreement value between the perturbations is obtained which leads to meta clusters in a hierarchical structure. The number of meta clusters is manually chosen based on the relative change in area under the cumulative distribution function curve. (Clustering Output, Section 1. Delta Area)
This clustering approach is fast and performs better than comparable techniques Weber et al. (2016).
First, group cells into clusters with FlowSOM using self-organizing map clustering and minimal spanning trees, and subsequently perform meta-clustering with ConsensusClusterPlus which determines cluster count and membership by stability evidence.
A self-organizing map (SOM) is an artificial neural network that reflects the topological information of the input in lower dimensions.
Meta clustering is carried out based on a consensus hierarchical method. By re-sampling from the original data and clustering the samples, an agreement value between the perturbations is obtained which leads to meta clusters in a hierarchical structure. The number of meta clusters is manually chosen based on the relative change in area under the cumulative distribution function curve. (Clustering Output, Section 1. Delta Area)
This clustering approach is fast and performs better than comparable techniques Weber et al. (2016).
Choose Clustering Parameters
Invalid clustering features
Please select more features.
No features in the current selection.
Invalid clustering dimensions
Need higher X/Y-dimension or lower maximum number of clusters.
The following has to be TRUE: floor(0.8*(xdim*ydim)) >= maxK.
Differential Marker Expression Analysis
Here, you can compute differential marker expression between your conditions. The boxplots can help to get a rough
overview of the different markers in order to see which markers might be differentially expressed between conditions.
There are various options for editing the boxplots (e.g. facet by antigen or cluster ID, color by condition, sample ID, ...).
There are two different analysis types:
Differential Marker Expression (sometimes called differential states) which is a differential analysis of marker expression per condition,
overall or cluster-wise. This analysis can be performed using
- limma: limma is a tool originally designed for detecting differential gene expression, here adapted for differential median marker expression (overall or per cluster). It fits a linear model and is very fast.
- a linear mixed effect model (LMM): LMMs allow for modelling correlations between different samples in the dataset, i.e. random effects. An example for a random effect is e.g. the patient ID when more than one sample was measured for each patient. When random effects are not specified, the model is equivalent to a linear model. With this method, median marker expression (overall or cluster-wise) is analyzed as well. Very fast.
- a model based on Earth Mover's Distance (CyEMD): With CyEMD, the overall distributions of marker expressions between conditions (overall or cluster-wise) are compared, not just the median marker expression. This is why this method takes longer but can yield more sensitive results.
- CytoGLMM: fits a generalized linear mixed model predicting the conditions from the whole expression vectors. Can only handle grouped data. Very fast.
- CytoGLM: fits a bootstrapped generalized linear model and can therefore be run without grouping variable. High runtime because of the bootstrap replications.
- Wilcoxon tests: When no grouping variable is specified, a Wilcoxon rank-sum test (= Mann-Whitney U test) is computed, otherwise, a Wilcoxon signed-rank test is performed. Very fast.
- t-test: Computes a paired or unpaired t-test. Very fast.
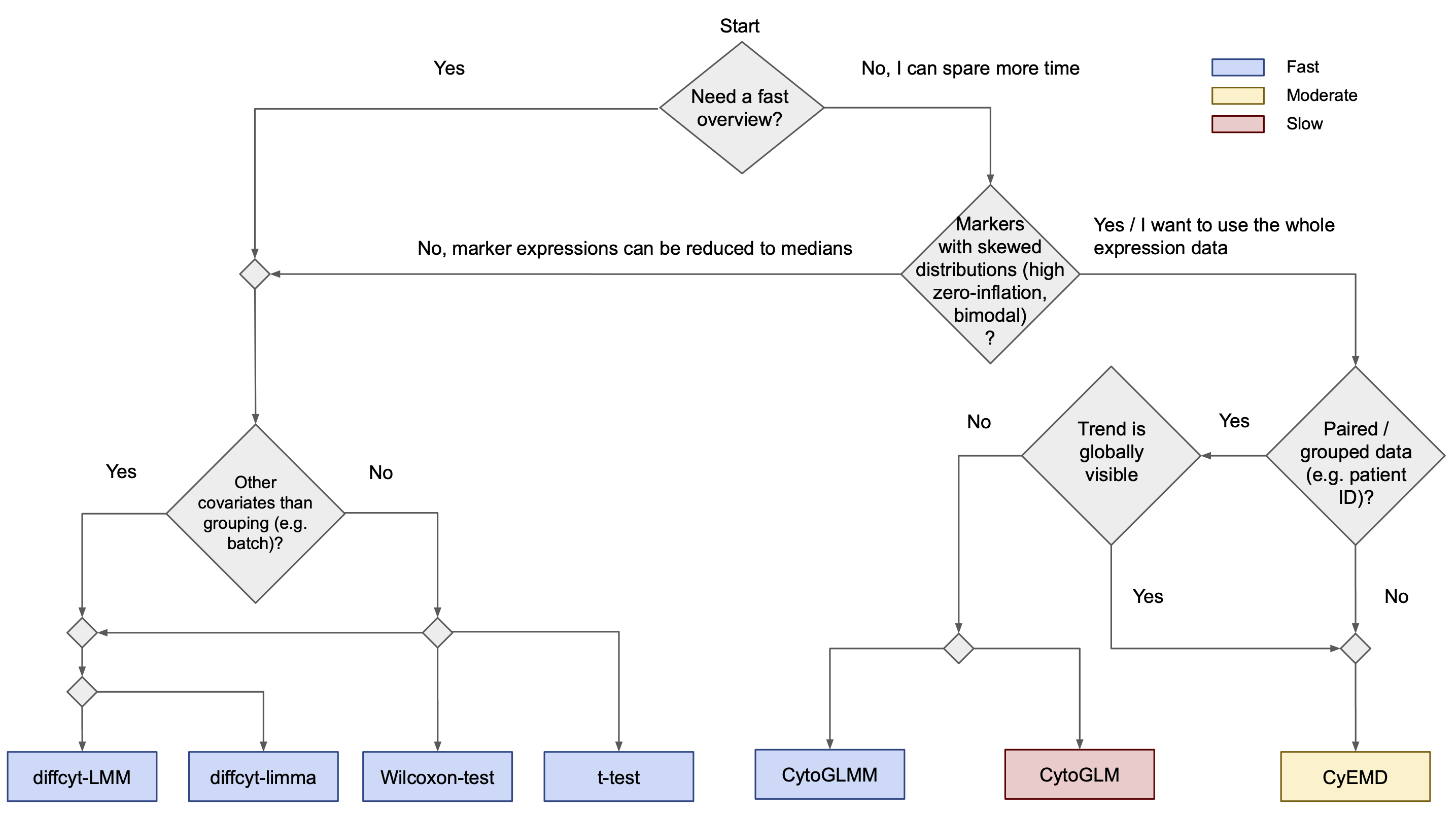
The second option is Differential Cluster Abundance (sometimes called differential abundance) compares the proportions of cell types across experimental
conditions per cluster and aims to highlight populations that are present at different ratios. The methods for this are
- edgeR: The edgeR package is used to fit models and calculate moderated tests at the cluster level. The statistical methods were originally designed for the analysis of gene expression data such as RNA-seq counts. Here, the methods are applied to cell counts.
- voom: Calculates tests for differential abundance of clusters, using functions from the limma package and voom method. Since count data are often heteroscedastic, the voom method is used to transform raw cluster cell counts and estimate observation-level weights to stabilize the mean-variance relationship.
- a generalized linear mixed model (GLMM): a generalized linear model is a flexible generalization of ordinary linear regression that allows for response variables that have error distribution models other than a normal distribution. A GLMM allows to include random effects like patient ID when more than one sample was measured per patient.
Boxplots
DE Method Comparison
Here, you can compare the results of different methods run on the same subset. Choose between Differential Cluster Abundance and Differential Marker Expression methods:
- Differential Cluster Abundance methods: edgeR, voom, GLMM
- Differential Marker Expression methods: limma, LMM, CyEMD, CytoGLMM, CytoGLM, Wilcoxon rank-sum test, Wilcoxon signed-rank test (for paired data), paired and unpaired t-test.
For more information, please refer to the DE analysis tab!
Choose Method and Parameters
Result Comparison
Loading...
Thanks for using our CyTOF Pipeline
We would like to thank you for using our App for analysing mass cytometry data. If you would like to save your results, click on the button on the top right. If you want to continue your analyses at a later point in time, you can upload the downloaded SCE object at the beginning of the workflow.
We would also be very pleased to receive your feedback on GitHub.
If you use our CyTOF pipeline for analysing your mass cytometry data please cite ...