Grand Forest • Supervised workflow user guide
Contents
1. Expression data format
Expression data must be uploaded as a data table in one of the formats described on the expression data file format page.
Besides the expression values, the data table must contain an extra column for the dependent variable. The type of the dependent variable depends on the type of model being trained:
- For classification/probability models the dependent variable must categorical. If the supplied values are numerical, they will be coerced into into class labels based on exact equality.
- For survival models the dependent variable must be a numerical survival time. Furthermore, and additional status variable must be supplied indicating patient status (
0= alive,1= dead). - For regression models the dependent variable must be numeric.
2. Training a model
In order to train a model, you must first upload a data file containing expression data and a dependent variable (phenotype). The data file should be formatted as described above. Once a file has been selected the upload will start automatically.
Next, we need to set the model parameters.
- Under Model type we have to choose the type of dependent variable we have in our data set. In this example we have class labels so we will select the Classification option.
- Under Dependent variable name we must specify the name of the column containing our dependent variable. In this example we input
"dmfs". The specified name must match the column name in the data file exactly, including capitalization. - Under Number of decision trees we can specify how many trees we want to train in our model. A large number of trees will increase the model stability but also the necessary computation time.
- Finally, under Network we need to select the network data source we want to use. By selecting Custom network we can also upload our own network file (see network file format for details). Let’s use only experimentally validated interactions from the IID database.

Once all parameters are set, click Train grand forest to train the model.
When the model is done training a brief summary will appear in the sidebar. Make sure to inspect this in order to spot potential problems. Under Genes in network we find the percentage of genes in the data set that were found in the network. If this number is very low it might suggest that the columns in the data file were not correct Entrez gene IDs.

Using the example data set
An example data set is also provided for demonstration. To use the example data, simply check the Use example data option from the sidebar.
The data set contains microarray data from 623 breast cancer samples compiled by Staiger et al. Each patient is annotated according to their five-year distance metastasis survival (DMFS) which is used as dependent variable when training the model.
3. Understanding the results
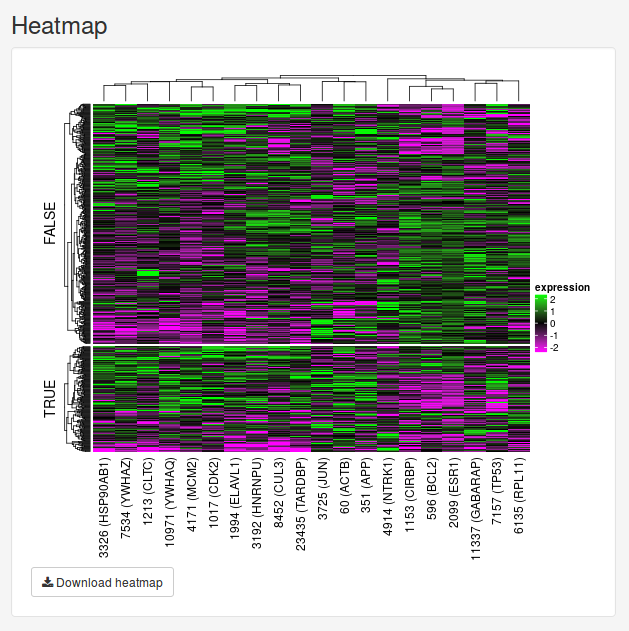
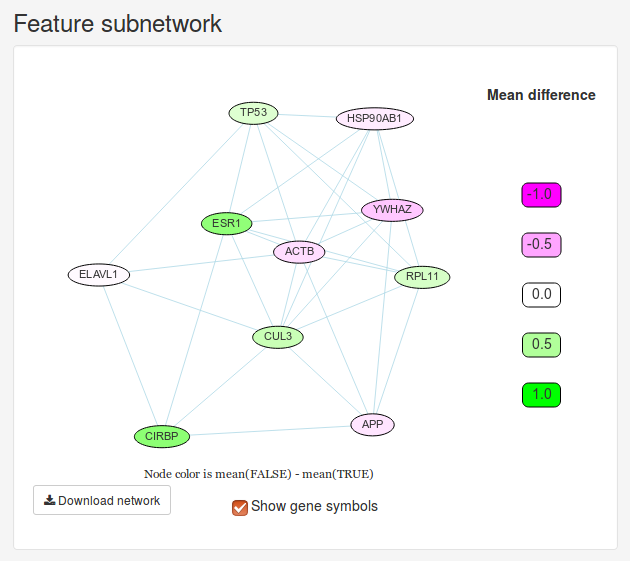
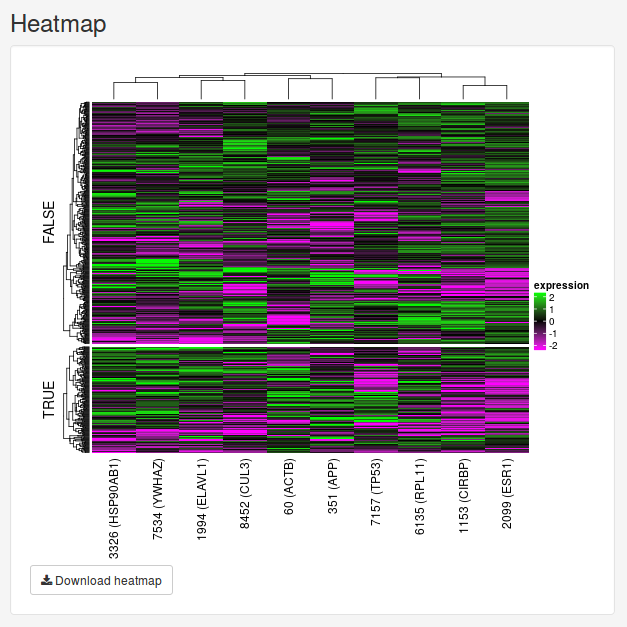
Once we have trained a model, we will initially be presented with a small network and a heatmap. These two figures visualize the 20 most important features determined by the model. The Feature subnetwork is constructed by extracting these genes from the source network along with all the interactions between them. If our data set only contains two classes, the nodes will also by colored according to the difference in mean expression between the two classes. The Feature heatmap shows the expression of all patients for the only top genes. For classification and probability models, the samples will be grouped based on the dependent variable.

We can change number of features we want to see using the slider in the sidebar under Number of top features to use. Let’s change it to 10 features instead.
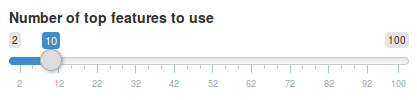
Once we move the slider, the feature subnetwork and heatmap will be updated automatically.
Gene set enrichment
Once we have found an interesting gene subnetwork, we can investigate whether these genes are significantly overrepresented in some GO terms, pathways or diseases. In order to perform gene set enrichment, select the Gene set enrichment tab above the feature table.
Under Enrichment type we can select which source of gene sets we want to check for enrichment. Here we select GO Biological process. The p-value cutoff and q-value cutoff parameters are used to filter results. Only gene sets with p- and q-value below cutoffs these will be reported.
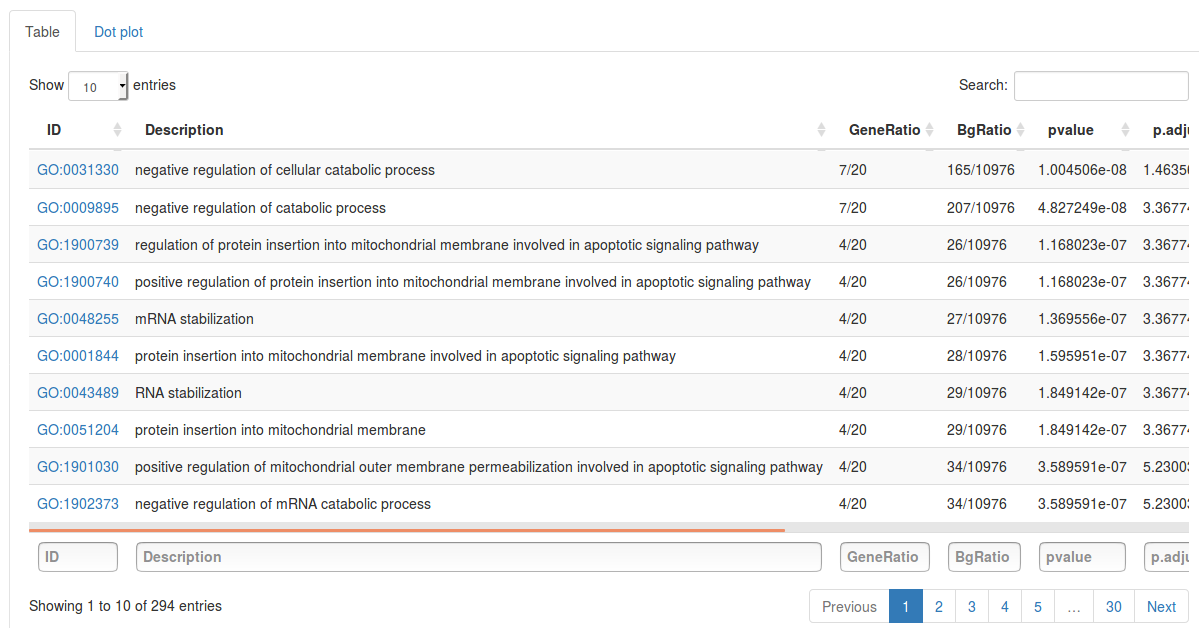
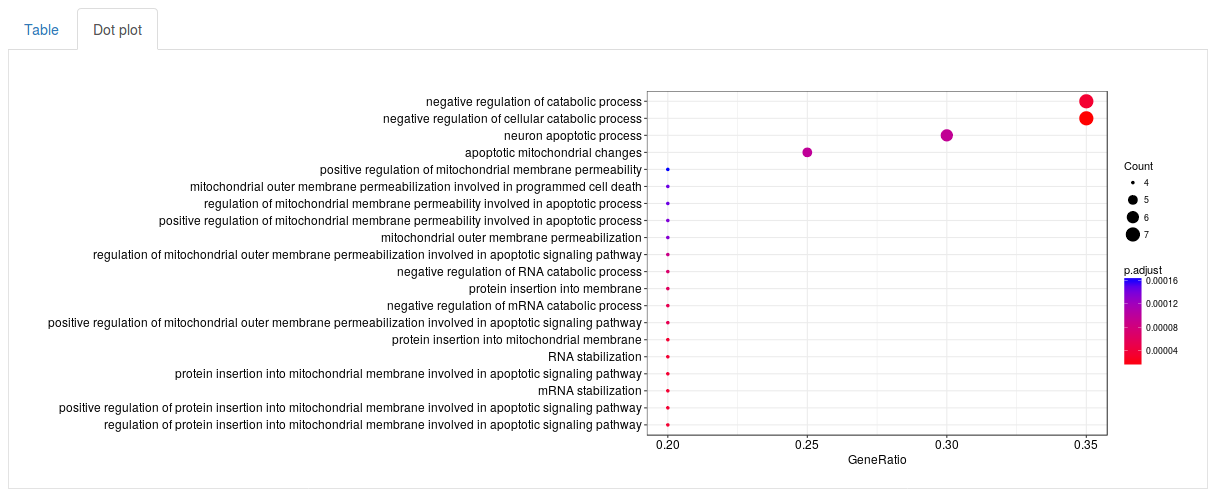
Click Run enrichment analysis to run the enrichment. After a little while a table will show up with all significantly enriched gene sets, ordered by p-value. For each gene set the table will show the name of the gene set (in this case GO term), how many genes in our subnetwork were actually found in that gene set, as well as a p-value, adjusted p-value and q-value. The search fields above the table can be used to search for specific gene sets. By selecting the Dot plot tab above the table we can also visualize the top results using a dot plot.
Finding drug and miRNA targets
We can also search for drugs and miRNAs targeting our genes of interest. First, select the Gene targets tab under Genes.
Under database we can choose the source database we want to search in. Here we select the DrugBank database to search for drugs targeting our top genes. Once you have selected a database click Get gene targets to start the search. The found targets will appear in a table below.
Each row in the table contains a gene and a drug targeting it. Click on the drug ID of a drug to open its page on the DrugBank website. Other columns will also link to external databases such as PubChem and KEGG if available. Using the search fields below the columns we can also filter the results by gene or drug name.
If we select the Network tab above we table we can also visualize the drug targets as a network. Here, each red node corresponds to a gene of interest and and each blue node corresponds to a drug.
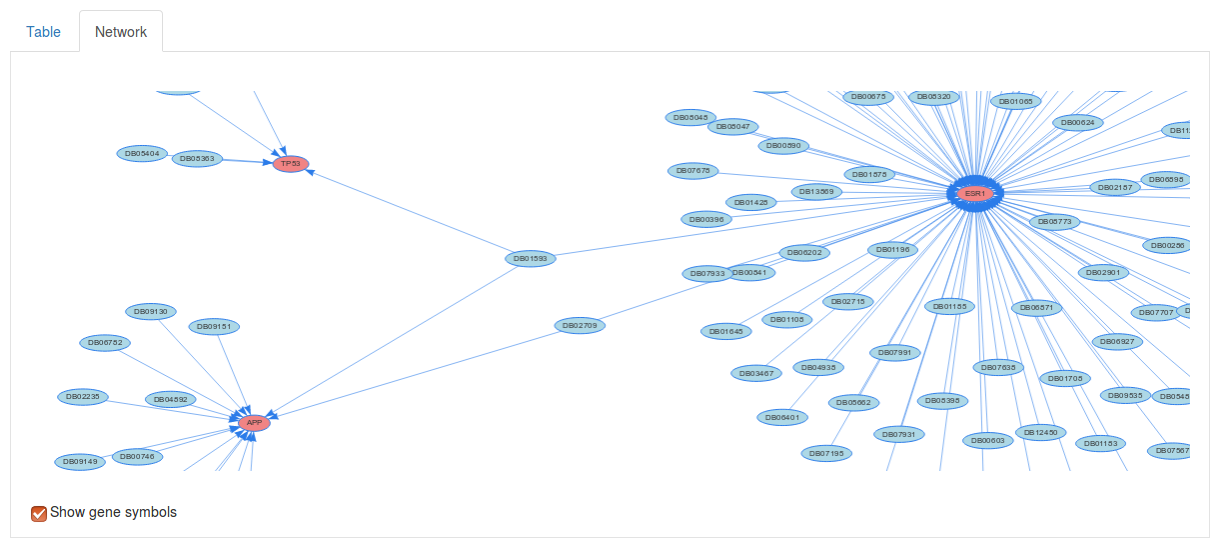
4. Model evaluation
In order to help get a sense of how well the method is able to model the data set, we provide a basic model evaluation through cross-validation. Select the Evaluation button at the top of the page to open the model evaluation page.
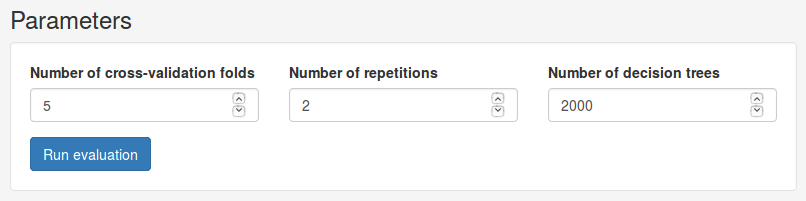
In the Parameters panel we can set basic parameters for the evaluation. Under Number of cross-validation folds we can choose how many folds we want to partition the data set into. We can also choose how many repetitions to perform and the number of trees to train. Here we will use 5 folds over 2 repetitions. Click Run evaluation to start the evaluation. Depending on the number of repetitions and decision trees we use, this may take a while.
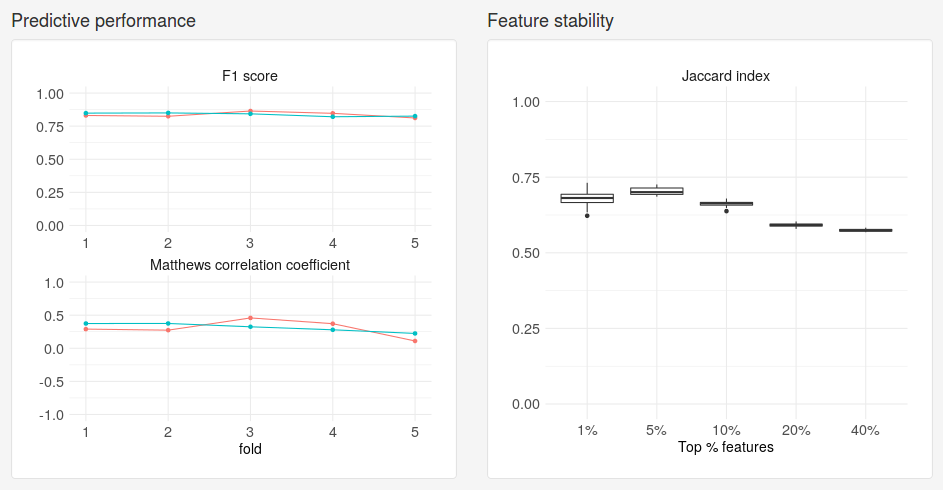
Once the evaluation is complete, we will be presented with two plots. Under Predictive performance we see how well the model predicted on the test set for each fold. The performance measures used depend on the type of model. For classification models performance will be reported as F1 score and Matthews correlation coefficient. Each dot corresponds to a different fold and each separate line corresponds to a repetition. Under Feature stability we see how much the models trained on different folds agreed on the most important genes for varying fractions of the full data set. The agreement is measured using the Jaccard index, where a value of 1 means perfect agreement and a value of 0 means no overlap.